Research
複雑な生命現象において、要素間の関係性(ネットワーク)や揺らぎ(個体差、偶然性)が、応答・情報伝達・進化といったシステム全体の振る舞いにどんな法則性を生むのかに関心を持って研究しています。
Network structure and control responses in chemical reaction networks
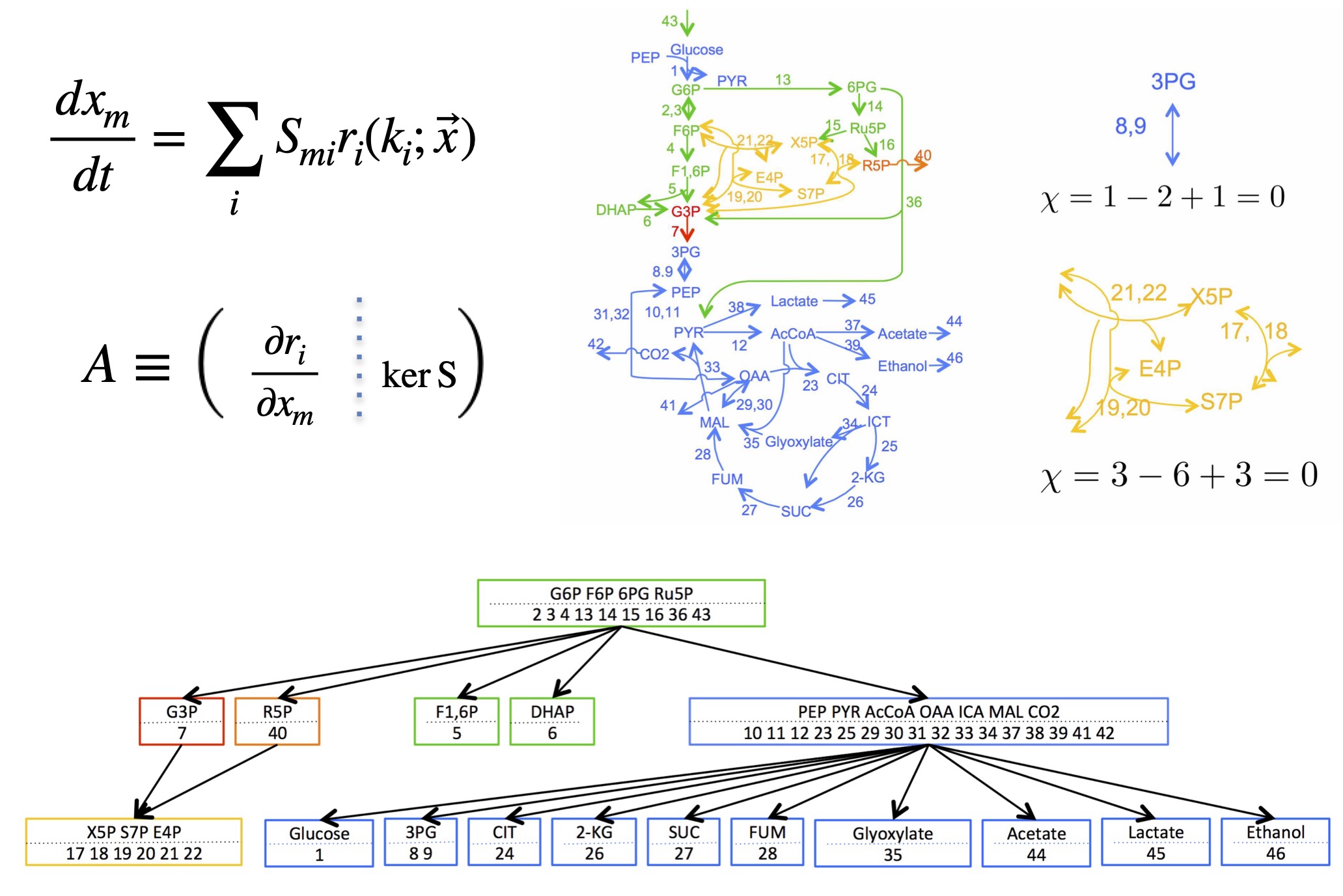
Inside cells, chemical reactions are connected by sharing the same chemical species (reactants and products), forming large-scale networks such as metabolic pathways.
In this research, we developed a theory that predicts—from the network connectivity alone—how metabolite concentrations and reaction fluxes at steady state change (i.e., sensitivities/responses) when enzyme activities are perturbed.
We found that nonzero responses exhibit two key properties:
Localization: For steady states, the effect of an enzyme perturbation does not spread across the entire network. Instead, it remains confined to a specific substructure (a buffering structure) characterized by a topological invariant analogous to the Euler characteristic: (# of species − # of reactions + # of independent cycles = 0).
Hierarchy: Buffering structures can be nested, which makes the influence propagate in a stepwise manner — “upstream → downstream”.
These results suggest that one reason biological systems are robust to external changes lies in the network structure itself.
We also showed that buffering structures govern not only perturbation responses but also bifurcation phenomena in chemical reaction networks (i.e., the diversity/multiplicity of steady states).
References
化学反応ネットワークのネットワーク構造と制御応答
細胞の中では、化学反応どうしが「同じ物質(反応物や生成物)を共有する」ことでつながっていて、代謝経路のような大きなネットワークを作っています。
この研究では、酵素の働きを少し変えたときに、物質の濃度や反応の流れ(フラックス)がどれくらい変化するか(=感度/応答)を、ネットワークのつながり方だけから予測できる理論を作りました。
その結果、変化が起きる場所(応答がゼロでない場所)の広がり方には、次の2つの特徴があることが分かりました。
局在化:定常状態に関して、酵素撹乱に対する影響はネットワーク全体に広がるわけではなく、オイラー数に類似した標数(分子の数ー反応の数+サイクルの数=0)をもつ特定の部分構造(緩衝構造)にとどまる。
階層性:緩衝構造が入れ子関係を形成することで、影響が「上流→下流」のように、段階的に伝わる
これらの結果は、生物が外からの変化に強い(頑健である)理由の一つが、ネットワークの構造そのものにあることを示唆しています。
また、緩衝構造は撹乱応答だけでなく、化学反応ネットワークにおける分岐現象(定常状態の多様性)を規定していることも分かりました。
参考URL / References
Interpreting evolutionary fluctuations: a new theory driven by skewed offspring distributions
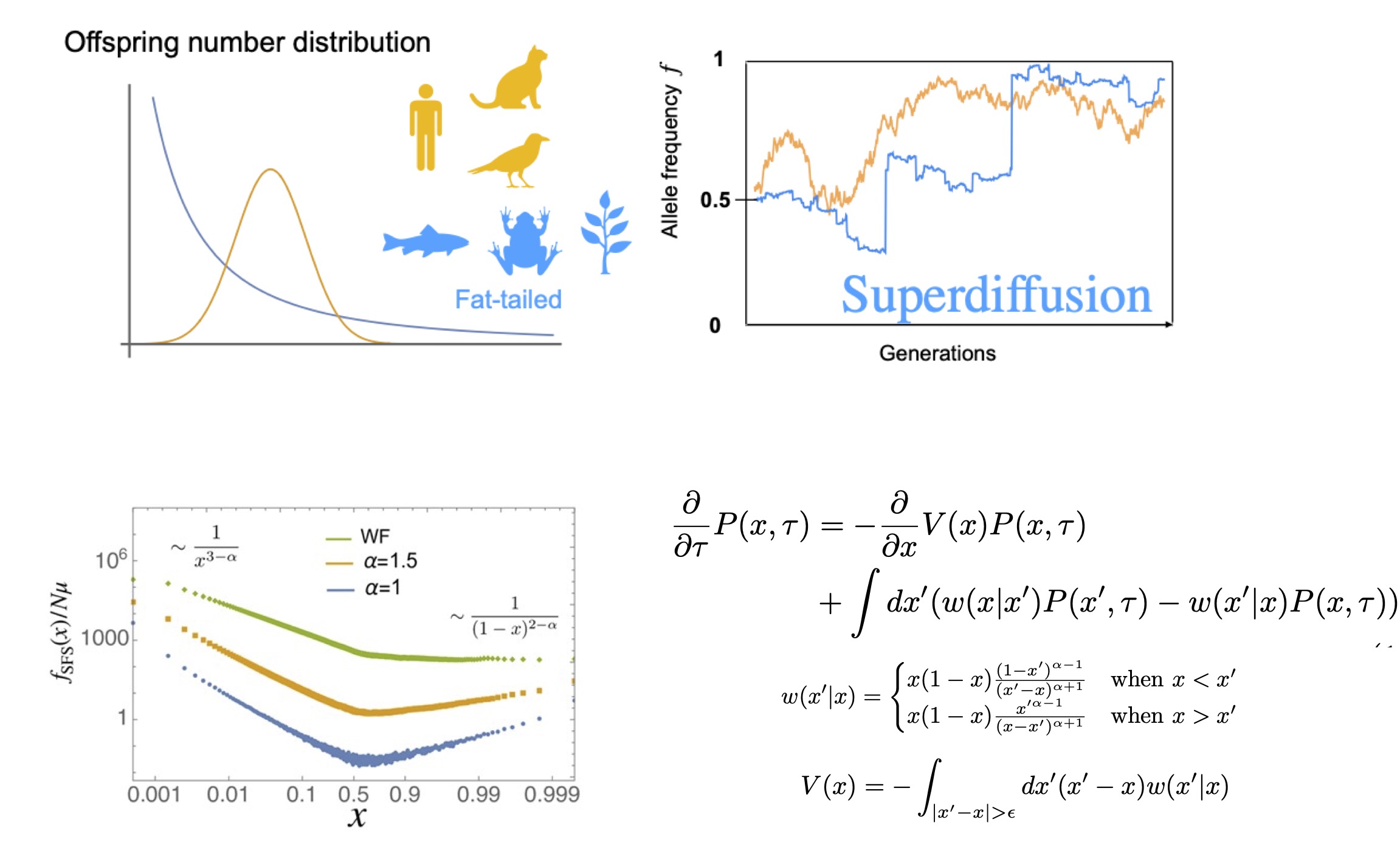
In many natural populations, individuals differ greatly in the number of offspring they leave. While most individuals leave few or no offspring, a small fraction can leave an exceptionally large number. In such situations, allele frequencies can fluctuate strongly due to chance—this is known as genetic drift.
However, classical population-genetic models often assume that the variance in offspring number is not too large, and therefore they cannot fully explain the large fluctuations observed in real populations.
In this work, using asymptotic analysis and scaling arguments, we showed that in populations with extremely broad offspring-number distributions, a natural bias against minority alleles can emerge.
This bias arises because the contribution of the “most prolific” individual in each generation fluctuates over time. As a result, this new bias competes with standard evolutionary forces such as selection and mutation, producing allele-frequency fluctuations that differ qualitatively from the classical picture.
Moreover, we derived simple scaling relations for the magnitude of allele-frequency fluctuations, fixation probabilities, times to extinction, and the resulting allele-frequency spectrum.
Such non-classical evolutionary laws are now beginning to be observed in fast-evolving populations such as microbes and viruses. Because these systems allow direct observation of evolutionary dynamics in laboratory evolution experiments, they offer promising opportunities to test theory–experiment connections.
References
進化の揺らぎを読み解く - 子孫数の偏りが生む新しい揺らぎの理論
自然界の多くの集団では、子どもの数が個体ごとに大きく異なることが知られています。 ほとんど子どもを残さない個体が多い一方で、ごく少数の個体が非常に多くの子どもを残す場合があります。こうした状況では、遺伝子の頻度が偶然によって大きく変動しやすくなります(これを遺伝的浮動と呼びます)。
しかし、これまでよく使われてきた集団遺伝学のモデルでは、「子どもの数のばらつきはそれほど大きくない」という仮定が置かれており、現実の集団で起きる大きな揺らぎを十分に説明できません。
過去をさかのぼる方向(どの個体が共通祖先か、など)については解析手法が発展していますが、時間を前に進めたとき(世代が進むとき)に、自然選択や突然変異と組み合わさって何が起きるのかについては、理論的・定量的な理解はまだ進んでいませんでした。
そこで本研究では、一般化中心極限定理とスケーリングの考え方を用いて、子どもの数のばらつきが非常に大きい集団では、少数派の遺伝子が不利になりやすい「バイアス」が自然に生じることを示しました。
このバイアスは、各世代で「最も多く子どもを残した個体」がどれくらい多かったか、という点が時間とともに変化することから生まれます。その結果、自然選択や突然変異といった通常の要因と、この新しいバイアスが競合し、遺伝子頻度が通常とは異なる仕方で揺らぐことが分かりました。
さらに、遺伝子頻度の揺らぎの大きさ、遺伝子が集団に定着する確率、絶滅までの時間、観測される遺伝子頻度分布といった性質を、シンプルな数式関係(スケーリング則)として整理しました。
このような新しい進化の法則性は、微生物やウイルスのように世代交代が速い生物集団でも、実際に観測され始めています。これらの生物は、実験室内で進化の過程を直接調べる進化実験に利用できるため、理論と実験を結びつけて検証できる点でも非常に有望です。
参考URL / References
Inferring Viral Transmission Pathways from Allele Frequency Time-Series Data
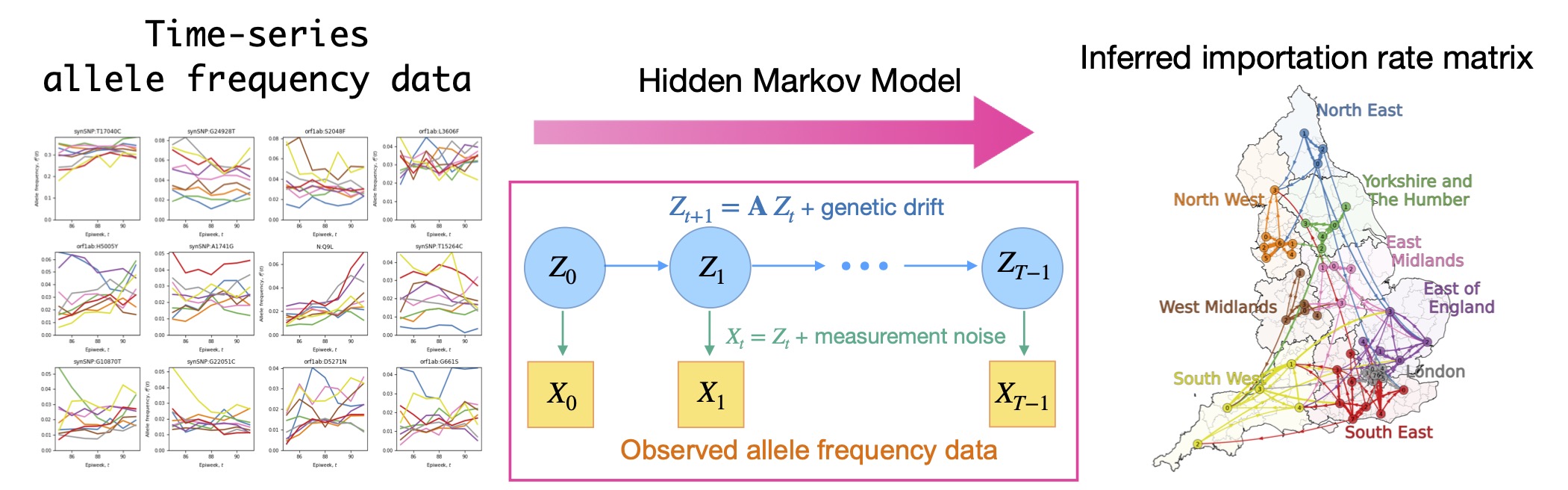
The COVID-19 pandemic has renewed awareness of the importance of interregional transmission, namely how pathogens spread across geographic regions. Traditionally, transmission has been inferred from human mobility data and contact histories; however, it has been difficult to capture rare transmission events occurring between socially or geographically distant regions.
In collaborative work with G. Isacchini, Q. Yu, and O. Hallatschek (University of California, Berkeley), we developed a mathematical method to directly infer importation transmission rates between regions from time-series data of allele frequencies. To account for observation errors and stochastic fluctuations in allele frequencies, we employed a hidden Markov model.
Applying this method to SARS-CoV-2 data revealed not only how interregional infection networks change across viral variants, but also the time scales over which transmission occurs and how transmission speeds differ between regions.
This study opens a new avenue for genome-based time-series analysis and is expected to contribute not only to future epidemiological monitoring and predictive models of pathogen spread, but also to a wide range of applications. The study was published in PNAS (2025) and also highlighted as a promising approach in a PNAS Commentary.
References
アレル頻度時系列データに基づくウイルス感染経路の推定
COVID-19のパンデミックは、病原体がどのように地域を越えて広がっていくのかという「地域間伝播」の重要性を再認識させました。従来は、人の移動データや接触履歴に基づいて伝播を推定してきましたが、社会的・地理的に離れた地域間で起きるような稀な伝播イベントを捉えることは困難でした。
米国カリフォルニア大学バークレー校 Giulio Isacchini研究員、米国ハーバード大学T.H.チャン公衆衛生大学院 QinQin Yu研究員、米国カリフォルニア大学バークレー校 Oskar Hallatschek 教授との共同研究で、SARS-CoV-2の豊富なゲノムデータに着目し、各地域のアレル頻度の時系列データから感染伝播の流入を直接推定する数理手法を開発しました。観測誤差やアレル頻度の揺らぎも考慮するために隠れマルコフモデルを利用しています。この手法を英国および米国のSARS-CoV-2データに適用した結果、変異株ごとに地域間の感染ネットワークがどのように変化するかに加えて、感染伝播がどの程度の時間スケールで起きているか、また、その速度が地域間でどのように異なるかといった動態も明らかになりました。本研究は、ゲノムデータに基づく新たな時系列解析の道を拓くものであり、今後の疫学モニタリングや病原体の拡散予測モデルへの応用だけでなく、さまざまなゲノムデータへの応用が期待されます。
本研究成果は、2025年11月26日に米国の国際学術誌「Proc. Natl. Acad. Sci. U.S.A」にオンライン掲載されました。また、Proc. Natl. Acad. Sci. U.S.AのCommentaryにも、有望な手法として取り上げられました。
参考URL / References
Information transmission in control networks
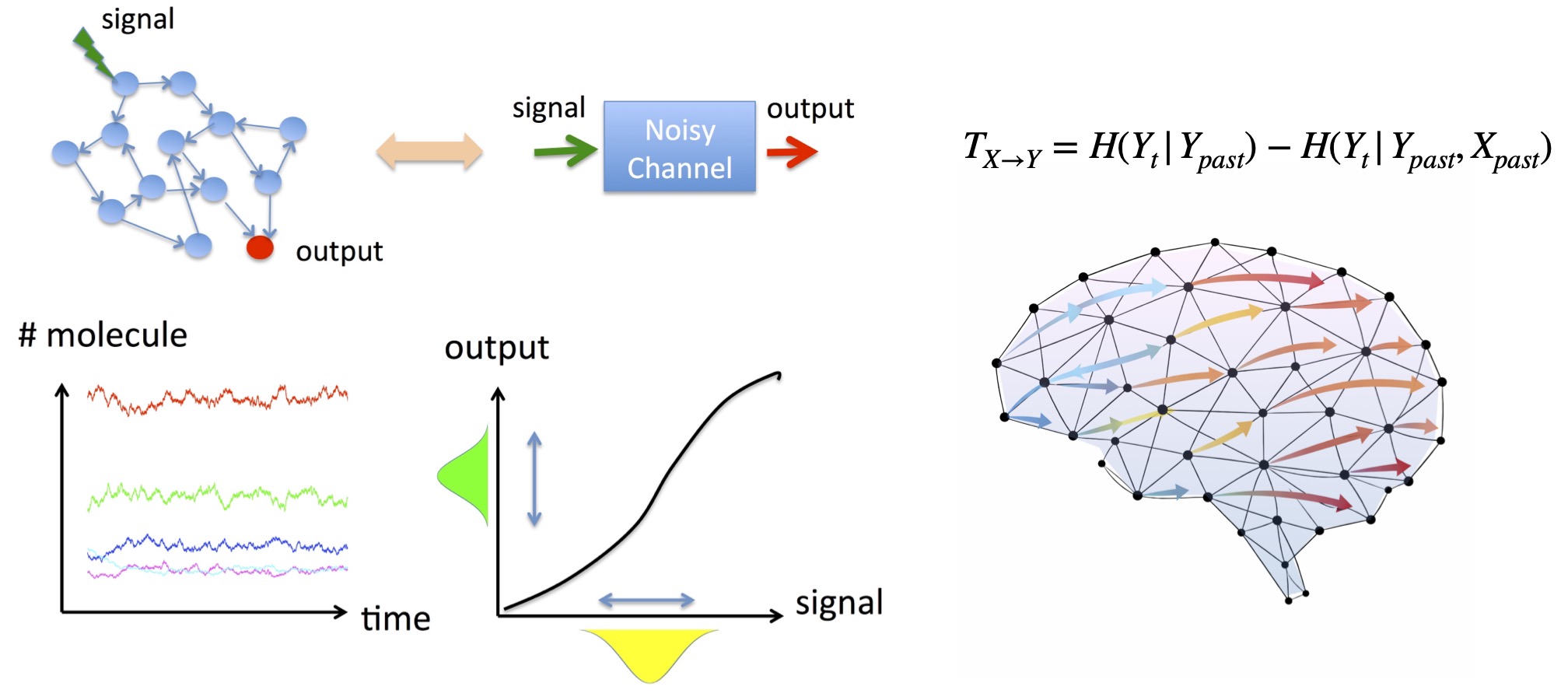
Accurate information transmission is a fundamental property shared by biological, social, and technological networks. Transfer entropy is a widely used measure that quantifies information flow, but even for small networks it often requires numerical computation, making it difficult to understand why information transmission works well.
In this research, focusing on stochastic Boolean networks, we derived a diagrammatic formula that allows transfer entropy to be computed analytically. The method applies to networks with arbitrary intermediate structure between an input signal and an output node, and also allows each node to implement a different logical function.
By decomposing transfer entropy into contributions from individual network components, we clarify which pathways carry information and provide design principles for network structures that maximize information transmission.
These results are applicable to understanding and designing real-world networks, from biological systems to engineered artificial networks. We are currently extending this theory to observational data and real networks, aiming to analyze information transmission directly from empirical measurements.
References
制御ネットワークにおける情報伝達
正確な情報伝達は、生物・社会・技術ネットワークに共通して重要な性質です。情報の流れを定量化する指標である移動エントロピー(Transfer Entropy)は、通常は小規模なネットワークであっても数値計算に頼る必要があり、なぜ情報がうまく伝わるのかという仕組みは分かりにくいままでした。
本研究では、確率的ブールネットワークを対象に、移動エントロピーを解析的に計算できる図式的な公式を導出しました。この解析手法は、入力から出力までの途中にあるネットワークが任意の構造を持ち、各ノードが異なる論理関数を持つような一般的場合にも適用できます。
移動エントロピーをネットワークの構成要素ごとに分解して表すことで、情報がどの経路を通って伝わるのかという仕組みを明確化するとともに、情報伝達を最大化するネットワーク構造の設計原理を与えます。この結果は、生物ネットワークや人工システムなど、実在するネットワークの理解や設計に応用可能です。
現在は、この理論を実際の観測データや実ネットワークに適用し、実データに基づいた情報伝達の解析へと研究を発展させています。